
Biomedical Computing
Biomedical computing combines the diagnostic and investigative aspects of biology and medical science with the power and problem-solving capabilities of modern computing. Computers are used to accelerate research learning, simulate patient behavior and visualize complex biological models.


Jeff Weiss
Computational Biomechanics
Orly Alter
Computational Biology
Tamara Bidone
Computational Models
Simulations of Biological Systems
Multi-Physics Models of Cancer Cells

Centers and Labs:
- Center for Integrative Biomedical Computing
- Muskuloskeletal Research Laboratory
- Genomic Signal Processing Lab
- Computational Biomechanics Group
Funded Research Projects:
Publications in Biomedical Computing:
 Scientific Visualization: Uncertainty, Multifield, Biomedical, and Scalable Visualization, C.D. Hansen, M. Chen, C.R. Johnson, A.E. Kaufman, H. Hagen (Eds.). Mathematics and Visualization, Springer, 2014. ISBN: 978-1-4471-6496-8 |
|
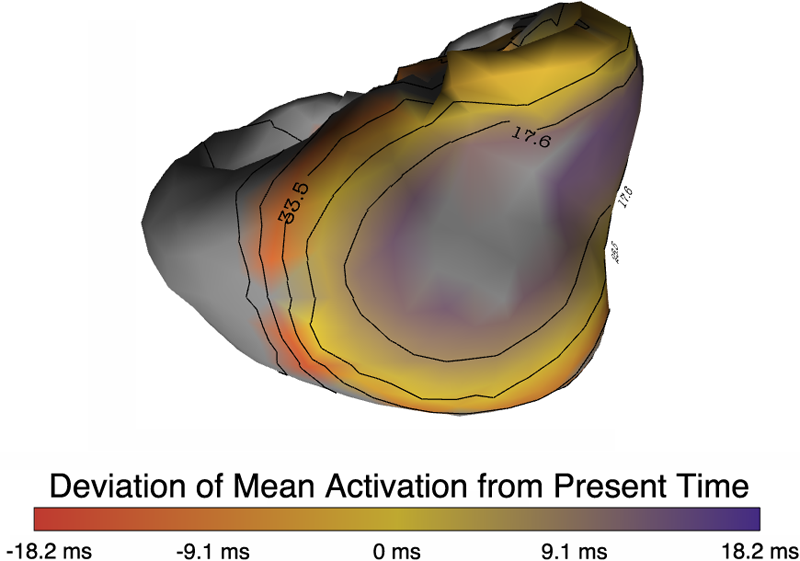
 Uncertainty Visualization in Forward and Inverse Cardiac Models B. Burton, B. Erem, K. Potter, P. Rosen, C.R. Johnson, D. Brooks, R.S. Macleod. In Computing in Cardiology CinC, pp. 57--60. 2013. ISSN: 2325-8861 Quantification and visualization of uncertainty in cardiac forward and inverse problems with complex geometries is subject to various challenges. Specific to visualization is the observation that occlusion and clutter obscure important regions of interest, making visual assessment difficult. In order to overcome these limitations in uncertainty visualization, we have developed and implemented a collection of novel approaches. To highlight the utility of these techniques, we evaluated the uncertainty associated with two examples of modeling myocardial activity. In one case we studied cardiac potentials during the repolarization phase as a function of variability in tissue conductivities of the ischemic heart (forward case). In a second case, we evaluated uncertainty in reconstructed activation times on the epicardium resulting from variation in the control parameter of Tikhonov regularization (inverse case). To overcome difficulties associated with uncertainty visualization, we implemented linked-view windows and interactive animation to the two respective cases. Through dimensionality reduction and superimposed mean and standard deviation measures over time, we were able to display key features in large ensembles of data and highlight regions of interest where larger uncertainties exist. |
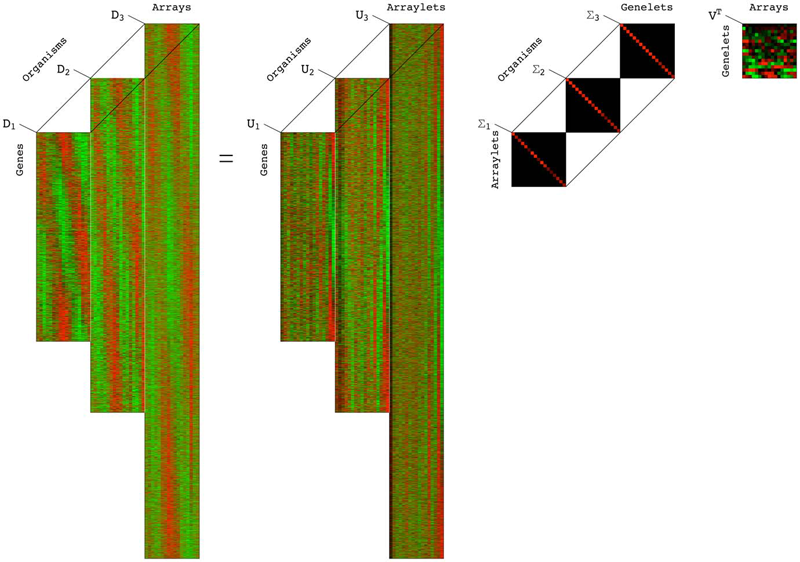
| SVD Identifies Transcript Length Distribution Functions from DNA Microarray Data and Reveals Evolutionary Forces Globally Affecting GBM Metabolism N.M. Bertagnolli, J.A. Drake, J.M. Tennessen, O. Alter. In Public Library of Science (PLoS) One, Vol. 8, No. 11, pp. article e78913. November, 2013. DOI: 10.1371/journal.pone.0078913 To search for evolutionary forces that might act upon transcript length, we use the singular value decomposition (SVD) to identify the length distribution functions of sets and subsets of human and yeast transcripts from profiles of mRNA abundance levels across gel electrophoresis migration distances that were previously measured by DNA microarrays. We show that the SVD identifies the transcript length distribution functions as “asymmetric generalized coherent states” from the DNA microarray data and with no a-priori assumptions. Comparing subsets of human and yeast transcripts of the same gene ontology annotations, we find that in both disparate eukaryotes, transcripts involved in protein synthesis or mitochondrial metabolism are significantly shorter than typical, and in particular, significantly shorter than those involved in glucose metabolism. Comparing the subsets of human transcripts that are overexpressed in glioblastoma multiforme (GBM) or normal brain tissue samples from The Cancer Genome Atlas, we find that GBM maintains normal brain overexpression of significantly short transcripts, enriched in transcripts that are involved in protein synthesis or mitochondrial metabolism, but suppresses normal overexpression of significantly longer transcripts, enriched in transcripts that are involved in glucose metabolism and brain activity. These global relations among transcript length, cellular metabolism and tumor development suggest a previously unrecognized physical mode for tumor and normal cells to differentially regulate metabolism in a transcript length-dependent manner. The identified distribution functions support a previous hypothesis from mathematical modeling of evolutionary forces that act upon transcript length in the manner of the restoring force of the harmonic oscillator. |

|
Graph Diffusion Distance: A Difference Measure for Weighted Graphs Based on the Graph Laplacian Exponential Kernel D.K. Hammond, Y. Gur, C.R. Johnson. In Proceedings of the IEEE global conference on information and signal processing (GlobalSIP'13), Austin, Texas, pp. 419--422. 2013. DOI: 10.1109/GlobalSIP.2013.6736904 We propose a novel difference metric, called the graph diffusion distance (GDD), for quantifying the difference between two weighted graphs with the same number of vertices. Our approach is based on measuring the average similarity of heat diffusion on each graph. We compute the graph Laplacian exponential kernel matrices, corresponding to repeatedly solving the heat diffusion problem with initial conditions localized to single vertices. The GDD is then given by the Frobenius norm of the difference of the kernels, at the diffusion time yielding the maximum difference. We study properties of the proposed distance on both synthetic examples, and on real-data graphs representing human anatomical brain connectivity. |
|
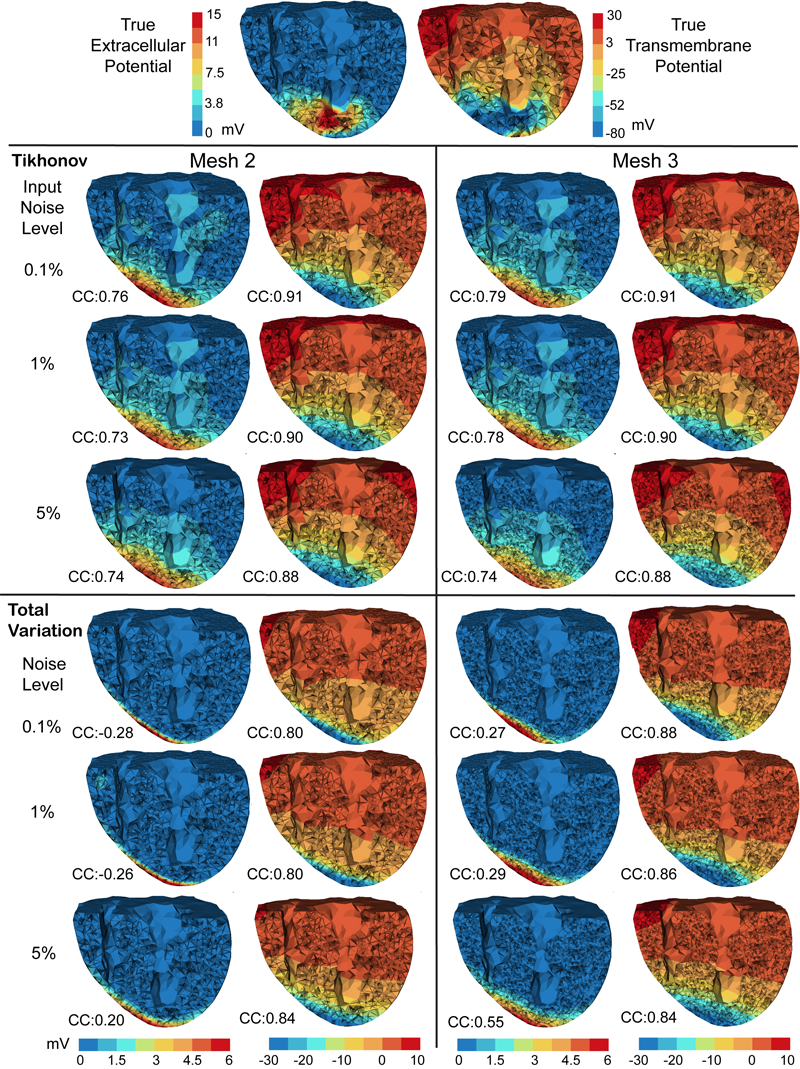
Inverse Electrocardiographic Source Localization of Ischemia: An Optimization Framework and Finite Element Solution D. Wang, R.M. Kirby, R.S. MacLeod, C.R. Johnson. In Journal of Computational Physics, Vol. 250, Academic Press, pp. 403--424. 2013. ISSN: 0021-9991 DOI: 10.1016/j.jcp.2013.05.027 With the goal of non-invasively localizing cardiac ischemic disease using bodysurface potential recordings, we attempted to reconstruct the transmembrane potential (TMP) throughout the myocardium with the bidomain heart model. The task is an inverse source problem governed by partial differential equations (PDE). Our main contribution is solving the inverse problem within a PDE-constrained optimization framework that enables various physically-based constraints in both equality and inequality forms. We formulated the optimality conditions rigorously in the continuum before deriving finite element discretization, thereby making the optimization independent of discretization choice. Such a formulation was derived for the L2-norm Tikhonov regularization and the total variation minimization. The subsequent numerical optimization was fulfilled by a primal-dual interior-point method tailored to our problem's specific structure. Our simulations used realistic, fiberincluded heart models consisting of up to 18,000 nodes, much finer than any inverse models previously reported. With synthetic ischemia data we localized ischemic regions with roughly a 10% false-negative rate or a 20% false-positive rate under conditions up to 5% input noise. With ischemia data measured from animal experiments, we reconstructed TMPs with roughly 0.9 correlation with the ground truth. While precisely estimating the TMP in general cases remains an open problem, our study shows the feasibility of reconstructing TMP during the ST interval as a means of ischemia localization. Keywords: cvrti, 2P41 GM103545-14 |
|
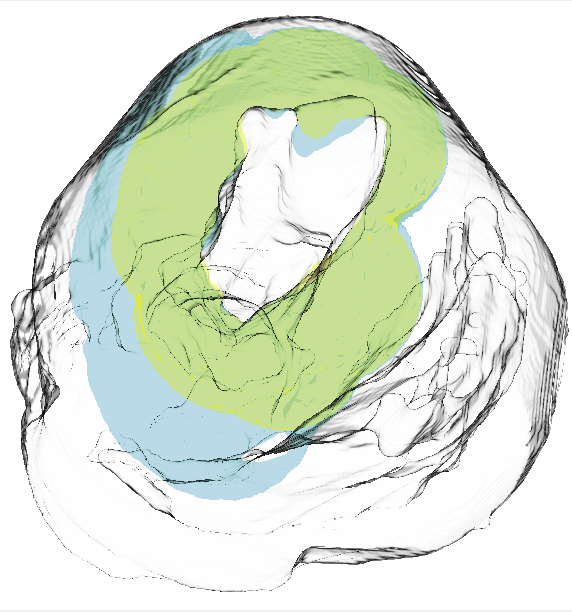
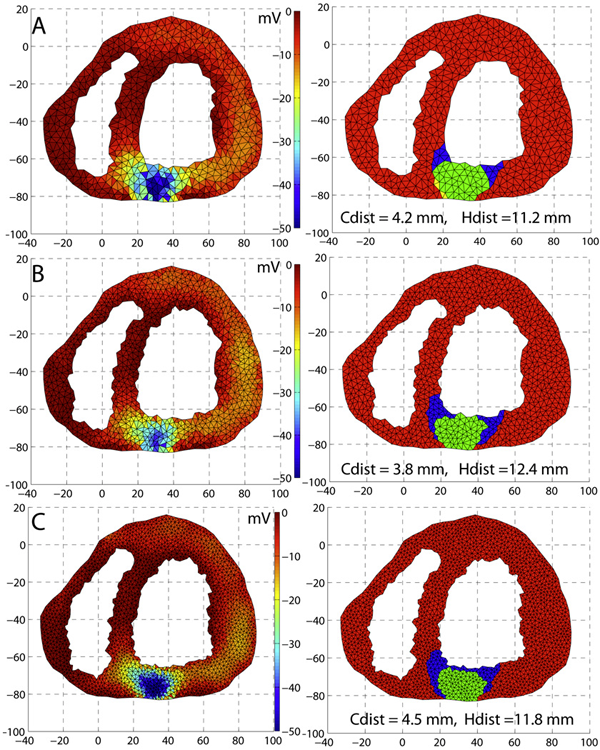
Visualization for understanding uncertainty in the simulation of myocardial ischemia P. Rosen, B. Burton, K. Potter, C.R. Johnson. In Proceedings of the 2013 Workshop on Visualization in Medicine and Life Sciences, 2013. We have created the Myocardial Uncertainty Viewer (muView) tool for exploring data stemming from the forward simulation of cardiac ischemia. The simulation uses a collection of conductivity values to understand how ischemic regions effect the undamaged anisotropic heart tissue. The data resulting from the simulation is multivalued and volumetric and thus, for every data point, we have a collection of samples describing cardiac electrical properties. muView combines a suite of visual analysis methods to explore the area surrounding the ischemic zone and identify how perturbations of variables changes the propagation of their effects. |
|
A Higher-Order Generalized Singular Value Decomposition for Comparison of Global mRNA Expression from Multiple Organisms S.P. Ponnapalli, M.A. Saunders, C.F. Van Loan, O. Alter. In PLoS One, Vol. 6, No. 12, pp. e28072. 2012. DOI: 10.1371/journal.pone.0028072 |
|
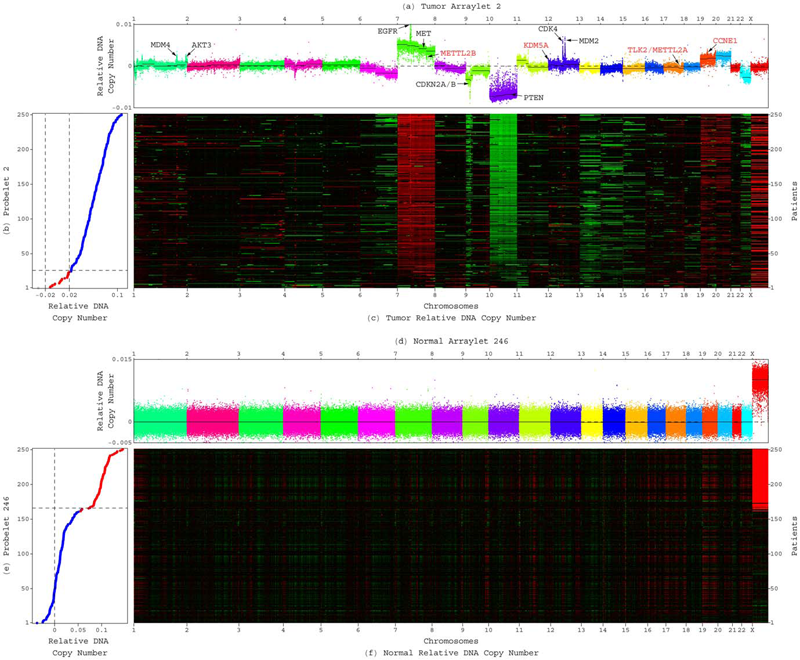
GSVD Comparison of Patient-Matched Normal and Tumor aCGH Profiles Reveals Global Copy-Number Alterations Predicting Glioblastoma Multiforme Survival C.H. Lee, B.O. Alpert, P. Sankaranarayanan, O. Alter. In PLoS ONE, Vol. 7, No. 1, Public Library of Science, pp. e30098. 2012. DOI: 10.1371/journal.pone.0030098 Despite recent large-scale profiling efforts, the best prognostic predictor of glioblastoma multiforme (GBM) remains the patient's age at diagnosis. We describe a global pattern of tumor-exclusive co-occurring copy-number alterations (CNAs) that is correlated, possibly coordinated with GBM patients' survival and response to chemotherapy. The pattern is revealed by GSVD comparison of patient-matched but probe-independent GBM and normal aCGH datasets from The Cancer Genome Atlas (TCGA). We find that, first, the GSVD, formulated as a framework for comparatively modeling two composite datasets, removes from the pattern copy-number variations (CNVs) that occur in the normal human genome (e.g., female-specific X chromosome amplification) and experimental variations (e.g., in tissue batch, genomic center, hybridization date and scanner), without a-priori knowledge of these variations. Second, the pattern includes most known GBM-associated changes in chromosome numbers and focal CNAs, as well as several previously unreported CNAs in greater than 3\% of the patients. These include the biochemically putative drug target, cell cycle-regulated serine/threonine kinase-encoding TLK2, the cyclin E1-encoding CCNE1, and the Rb-binding histone demethylase-encoding KDM5A. Third, the pattern provides a better prognostic predictor than the chromosome numbers or any one focal CNA that it identifies, suggesting that the GBM survival phenotype is an outcome of its global genotype. The pattern is independent of age, and combined with age, makes a better predictor than age alone. GSVD comparison of matched profiles of a larger set of TCGA patients, inclusive of the initial set, confirms the global pattern. GSVD classification of the GBM profiles of an independent set of patients validates the prognostic contribution of the pattern. |
|
An optimization framework for inversely estimating myocardial transmembrane potentials and localizing ischemia D. Wang, R.M. Kirby, R.S. Macleod, C.R. Johnson. In Proceedings of the International Conference of the IEEE Engineering in Medicine and Biology Society (EMBS), pp. 1680--1683. 2011. DOI: 10.1109/IEMBS.2011.6090483 PubMed ID: 22254648 PubMed Central ID: PMC3336368 By combining a static bidomain heart model with a torso conduction model, we studied the inverse electrocardiographic problem of computing the transmembrane potentials (TMPs) throughout the myocardium from a body-surface potential map, and then used the recovered potentials to localize myocardial ischemia. Our main contribution is solving the inverse problem within a constrained optimization framework, which is a generalization of previous methods for calculating transmembrane potentials. The framework offers ample flexibility for users to apply various physiologically-based constraints, and is well supported by mature algorithms and solvers developed by the optimization community. By avoiding the traditional inverse ECG approach of building the lead-field matrix, the framework greatly reduces computation cost and, by setting the associated forward problem as a constraint, the framework enables one to flexibly set individualized resolutions for each physical variable, a desirable feature for balancing model accuracy, ill-conditioning and computation tractability. Although the task of computing myocardial TMPs at an arbitrary time instance remains an open problem, we showed that it is possible to obtain TMPs with moderate accuracy during the ST segment by assuming all cardiac cells are at the plateau phase. Moreover, the calculated TMPs yielded a good estimate of ischemic regions, which was of more clinical interest than the voltage values themselves. We conducted finite element simulations of a phantom experiment over a 2D torso model with synthetic ischemic data. Preliminary results indicated that our approach is feasible and suitably accurate for the common case of transmural myocardial ischemia. |
|
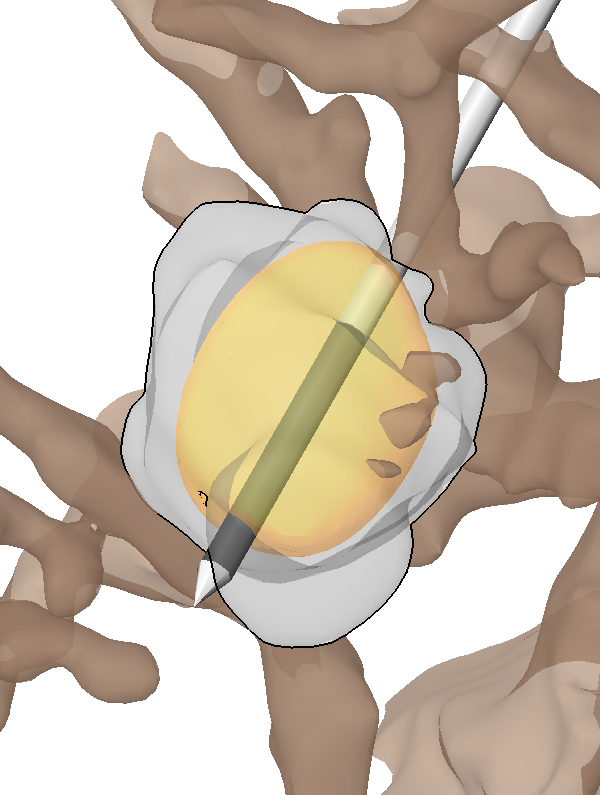
Sensitivity Analysis for the Optimization of Radiofrequency Ablation in the Presence of Material Parameter Uncertainty I. Altrogge, T. Preusser, T. Kroeger, S. Haase, T. Paetz, R.M. Kirby. In International Journal for Uncertainty Quantification, 2011. We present a sensitivity analysis of the optimization of the probe placement in radiofrequency (RF) ablation which takes the uncertainty associated with bio-physical tissue properties (electrical and thermal conductivity) into account. Our forward simulation of RF ablation is based upon a system of partial differential equations (PDEs) that describe the electric potential of the probe and the steady state of the induced heat. The probe placement is optimized by minimizing a temperature-based objective function such that the volume of destroyed tumor tissue is maximized. The resulting optimality system is solved with a multi-level gradient descent approach. By evaluating the corresponding optimality system for certain realizations of tissue parameters (i.e. at certain, well-chosen points in the stochastic space) the sensitivity of the system can be analyzed with respect to variations in the tissue parameters. For the interpolation in the stochastic space we use a stochastic finite element approach with piecewise multilinear ansatz functions on adaptively refined, hierarchical grids. We underscore the significance of the approach by applying the optimization to CT data obtained from a real RF ablation case. Keywords: netl, stochastic sensitivity analysis, stochastic partial di erential equations, stochastic nite element method, adaptive sparse grid, heat transfer, multiscale modeling, representation of uncertainty |
| Cardiac Position Sensitivity Study in the Electrocardiographic Forward Problem Using Stochastic Collocation and Boundary Element Methods D.J. Swenson, S.E. Geneser, J.G. Stinstra, R.M. Kirby, R.S. MacLeod. In Annals of Biomedical Engineering, Vol. 39, No. 12, pp. 2900--2910. 2011. DOI: 10.1007/s10439-011-0391-5 PubMed ID: 21909818 PubMed Central ID: PMC336204 The electrocardiogram (ECG) is ubiquitously employed as a diagnostic and monitoring tool for patients experiencing cardiac distress and/or disease. It is widely known that changes in heart position resulting from, for example, posture of the patient (sitting, standing, lying) and respiration significantly affect the body-surface potentials; however, few studies have quantitatively and systematically evaluated the effects of heart displacement on the ECG. The goal of this study was to evaluate the impact of positional changes of the heart on the ECG in the specific clinical setting of myocardial ischemia. To carry out the necessary comprehensive sensitivity analysis, we applied a relatively novel and highly efficient statistical approach, the generalized polynomial chaos-stochastic collocation method, to a boundary element formulation of the electrocardiographic forward problem, and we drove these simulations with measured epicardial potentials from whole-heart experiments. Results of the analysis identified regions on the body-surface where the potentials were especially sensitive to realistic heart motion. The standard deviation (STD) of ST-segment voltage changes caused by the apex of a normal heart, swinging forward and backward or side-to-side was approximately 0.2 mV. Variations were even larger, 0.3 mV, for a heart exhibiting elevated ischemic potentials. These variations could be large enough to mask or to mimic signs of ischemia in the ECG. Our results suggest possible modifications to ECG protocols that could reduce the diagnostic error related to postural changes in patients possibly suffering from myocardial ischemia. |
| Minimum Information about a Cardiac Electrophysiology Experiment (MICEE): Standardised reporting for model reproducibility, interoperability, and data sharing T.A. Quinn, S. Granite, M.A. Allessie, C. Antzelevitch, C. Bollensdorff, G. Bub, R.A.B. Burton, E. Cerbai, P.S. Chen, M. Delmar, D. DiFrancesco, Y.E. Earm, I.R. Efimov, M. Egger, E. Entcheva, M. Fink, R. Fischmeister, M.R. Franz, A. Garny, W.R. Giles, T. Hannes, S.E. Harding, P.J. Hunter, s, G. Iribe, J. Jalife, C.R. Johnson, R.S. Kass, I. Kodama, G. Koren, P. Lord, V.S. Markhasin, S. Matsuoka, A.D. McCulloch, G.R. Mirams, G.E. Morley, S. Nattel, D. Noble, S.P. Olesen, A.V. Panfilov, N.A. Trayanova, U. Ravens, S. Richard, D.S. Rosenbaum, Y. Rudy, F. Sachs, F.B. Sachse, D.A. Saint, U. Schotten, O. Solovyova, P. Taggart, L. Tung, A. Varrò, P.G. Volders, K. Wang, J.N. Weiss, E. Wettwer, E. White, R. Wilders, R.L. Winslow, P. Kohl. In Progress in Biophysics and Molecular Biology, Vol. 107, No. 1, Elsevier, pp. 4--10. October, 2011. DOI: 10.1016/j.pbiomolbio.2011.07.001 PubMed Central ID: PMC3190048 Cardiac experimental electrophysiology is in need of a well-defined Minimum Information Standard for recording, annotating, and reporting experimental data. As a step toward establishing this, we present a draft standard, called Minimum Information about a Cardiac Electrophysiology Experiment (MICEE). The ultimate goal is to develop a useful tool for cardiac electrophysiologists which facilitates and improves dissemination of the minimum information necessary for reproduction of cardiac electrophysiology research, allowing for easier comparison and utilisation of findings by others. It is hoped that this will enhance the integration of individual results into experimental, computational, and conceptual models. In its present form, this draft is intended for assessment and development by the research community. We invite the reader to join this effort, and, if deemed productive, implement the Minimum Information about a Cardiac Electrophysiology Experiment standard in their own work. Keywords: Minimum Information Standard; Cardiac electrophysiology; Data sharing; Reproducibility; Integration; Computational modelling |
| Quantifying variability in radiation dose due to respiratory-induced tumor motion S.E. Geneser, J.D. Hinkle, R.M. Kirby, Bo Wang, B. Salter, S. Joshi. In Medical Image Analysis, Vol. 15, No. 4, pp. 640--649. 2011. DOI: 10.1016/j.media.2010.07.003 |
|
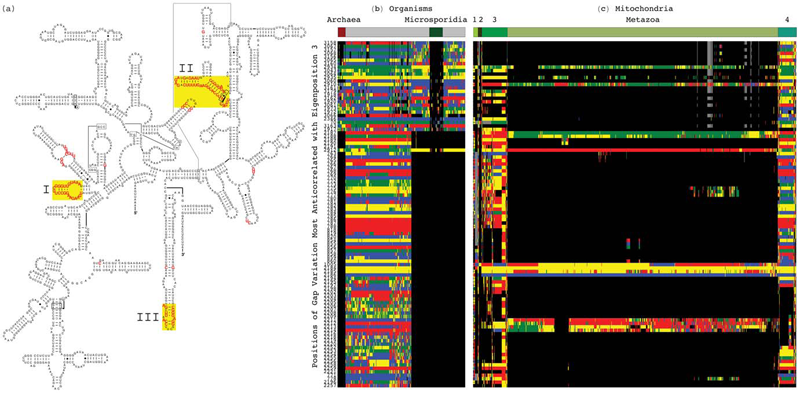
Tensor Decomposition Reveals Concurrent Evolutionary Convergences and Divergences and Correlations with Structural Motifs in Ribosomal RNA C. Muralidhara, A.M. Gross, R.R. Gutell, O. Alter. In PLoS ONE, Vol. 6, No. 4, Public Library of Science, pp. e18768. April, 2011. DOI: 10.1371/journal.pone.0018768 Evolutionary relationships among organisms are commonly described by using a hierarchy derived from comparisons of ribosomal RNA (rRNA) sequences. We propose that even on the level of a single rRNA molecule, an organism's evolution is composed of multiple pathways due to concurrent forces that act independently upon different rRNA degrees of freedom. Relationships among organisms are then compositions of coexisting pathway-dependent similarities and dissimilarities, which cannot be described by a single hierarchy. We computationally test this hypothesis in comparative analyses of 16S and 23S rRNA sequence alignments by using a tensor decomposition, i.e., a framework for modeling composite data. Each alignment is encoded in a cuboid, i.e., a third-order tensor, where nucleotides, positions and organisms, each represent a degree of freedom. A tensor mode-1 higher-order singular value decomposition (HOSVD) is formulated such that it separates each cuboid into combinations of patterns of nucleotide frequency variation across organisms and positions, i.e., \"eigenpositions\" and corresponding nucleotide-specific segments of \"eigenorganisms,\" respectively, independent of a-priori knowledge of the taxonomic groups or rRNA structures. We find, in support of our hypothesis that, first, the significant eigenpositions reveal multiple similarities and dissimilarities among the taxonomic groups. Second, the corresponding eigenorganisms identify insertions or deletions of nucleotides exclusively conserved within the corresponding groups, that map out entire substructures and are enriched in adenosines, unpaired in the rRNA secondary structure, that participate in tertiary structure interactions. This demonstrates that structural motifs involved in rRNA folding and function are evolutionary degrees of freedom. Third, two previously unknown coexisting subgenic relationships between Microsporidia and Archaea are revealed in both the 16S and 23S rRNA alignments, a convergence and a divergence, conferred by insertions and deletions of these motifs, which cannot be described by a single hierarchy. This shows that mode-1 HOSVD modeling of rRNA alignments might be used to computationally predict evolutionary mechanisms. |
|
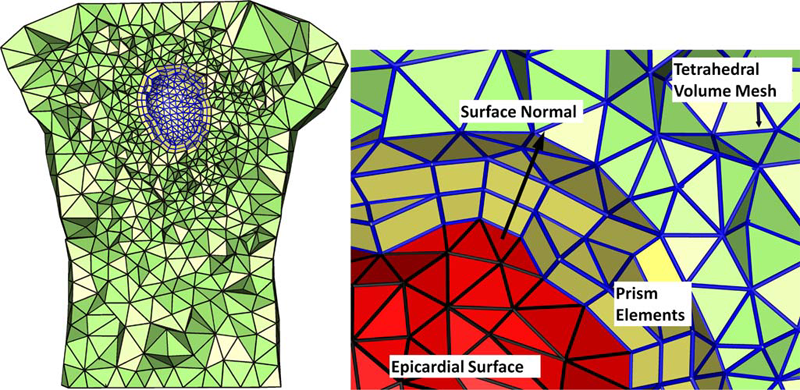
Finite Element Based Discretization and Regularization Strategies for 3D Inverse Electrocardiography D. Wang, R.M. Kirby, C.R. Johnson. In IEEE Transactions for Biomedical Engineering, Vol. 58, No. 6, pp. 1827--1838. 2011. PubMed ID: 21382763 PubMed Central ID: PMC3109267 We consider the inverse electrocardiographic problem of computing epicardial potentials from a body-surface potential map. We study how to improve numerical approximation of the inverse problem when the finite-element method is used. Being ill-posed, the inverse problem requires different discretization strategies from its corresponding forward problem. We propose refinement guidelines that specifically address the ill-posedness of the problem. The resulting guidelines necessitate the use of hybrid finite elements composed of tetrahedra and prism elements. Also, in order to maintain consistent numerical quality when the inverse problem is discretized into different scales, we propose a new family of regularizers using the variational principle underlying finite-element methods. These variational-formed regularizers serve as an alternative to the traditional Tikhonov regularizers, but preserves the L2 norm and thereby achieves consistent regularization in multiscale simulations. The variational formulation also enables a simple construction of the discrete gradient operator over irregular meshes, which is difficult to define in traditional discretization schemes. We validated our hybrid element technique and the variational regularizers by simulations on a realistic 3-D torso/heart model with empirical heart data. Results show that discretization based on our proposed strategies mitigates the ill-conditioning and improves the inverse solution, and that the variational formulation may benefit a broader range of potential-based bioelectric problems. |
| A New Family of Variational-Form-Based Regularizers for Reconstructing Epicardial Potentials from Body-Surface Mapping D.F. Wang, R.M. Kirby, R.S. MacLeod, C.R. Johnson. In Computing in Cardiology, 2010, pp. 93--96. 2010. |
| Effects of idealized joint geometry on finite element predictions of cartilage contact stresses in the hip A.E. Anderson, B.J. Ellis, S.A. Maas, J.A. Weiss. In Journal of Biomechanics, Vol. 43, No. 7, pp. 1351--1357. May, 2010. Computational models may have the ability to quantify the relationship between hip morphology, cartilage mechanics and osteoarthritis. Most models have assumed the hip joint to be a perfect ball and socket joint and have neglected deformation at the bone-cartilage interface. The objective of this study was to analyze finite element (FE) models of hip cartilage mechanics with varying degrees of simplified geometry and a model with a rigid bone material assumption to elucidate the effects on predictions of cartilage stress. A previously validated subject-specific FE model of a cadaveric hip joint was used as the basis for the models. Geometry for the bone-cartilage interface was either: (1) subject-specific (i.e. irregular), (2) spherical, or (3) a rotational conchoid. Cartilage was assigned either a varying (irregular) or constant thickness (smoothed). Loading conditions simulated walking, stair-climbing and descending stairs. FE predictions of contact stress for the simplified models were compared with predictions from the subject-specific model. Both spheres and conchoids provided a good approximation of native hip joint geometry (average fitting error ∼0.5 mm). However, models with spherical/conchoid bone geometry and smoothed articulating cartilage surfaces grossly underestimated peak and average contact pressures (50% and 25% lower, respectively) and overestimated contact area when compared to the subject-specific FE model. Models incorporating subject-specific bone geometry with smoothed articulating cartilage also underestimated pressures and predicted evenly distributed patterns of contact. The model with rigid bones predicted much higher pressures than the subject-specific model with deformable bones. The results demonstrate that simplifications to the geometry of the bone-cartilage interface, cartilage surface and bone material properties can have a dramatic effect on the predicted magnitude and distribution of cartilage contact pressures in the hip joint. Keywords: mrl |
| Resolution Strategies for the Finite-Element-Based Solution of the ECG Inverse Problem D.F. Wang, R.M. Kirby, C.R. Johnson. In IEEE Transactions on Biomedical Engineering, Vol. 57, No. 2, pp. 220--237. February, 2010. |
| Using the stochastic collocation method for the uncertainty quantification of drug concentration due to depot shape variability J.S. Preston, T. Tasdizen, C.M. Terry, A.K. Cheung, R.M. Kirby. In IEEE Transactions on Biomedical Engineering, Vol. 56, No. 3, Note: Epub 2008 Dec 2, pp. 609--620. 2009. PubMed ID: 19272865 |
| Global Effects of DNA Replication and DNA Replication Origin Activity on Eukaryotic Gene Expression, L. Omberg, J.R. Meyerson, K. Kobayashi, L.S. Drury, J.F.X. Diffley, O. Alter. In Nature Molecular Systems Biology, Vol. 5, No. 312, pp. (published online). October, 2009. DOI: 10.1038/msb.2009.70 |